tags:
- ngs
- fastq
type:
- course
class: m2ggb
subject:
- bioinformatics
Year: 2023yaml1 FastQ Files
contact: ludwig@univ-brest.fr
Copy the archive containing the data.
cd
cp /DATA/bioinfo/data.tar ~
tar xvf data.tar
This archive contains (among other) the FastQ files for 3 individuals
- father
- mother
- child
Paired-end sequencing implies that each individuals has 2 FastQ files
- xxxx.R1.fastq.gz for the Forward Reads
- xxxx.R2.fastq.gz for the matching Reverse Reads
The .gz extension indicates that the file has been compressed
The files are :
child.R1.fastq.gz
child.R2.fastq.gz
father.R1.fastq.gz
father.R2.fastq.gz
mother.R1.fastq.gz
mother.R2.fastq.gz
1.1. File Format
FastQ files are text file (human readable). It includes the reads coming from the sequencer. Each read is composed of 4 lines :
- starts with a
@symbol, followed by a sequence identifier. - the actual biological sequence.
- a
+symbol - the quality scores corresponding to the bases in Line 2.
1.1.1. Count the lines in the R1 file for the child. Check that it is a multiple of 4.
zcat DATA/child.R1.fastq.gz | wc -l
bash1.1.2. Count the lines in the R2 file for the child. Is it equal or different ? Why ?
zcat DATA/child.R2.fastq.gz | wc -l
bashFastQ files are a succession of 4 lines groups :
id1
seq1
+
qual1
id2
seq2
+
qual2
...
idn
seqn
+
qualn
by using paste - - - - we can group data into 4 columns :
id1 seq1 + qual1
id2 seq2 + qual2
...
idn seqn + qualn
The command cut -f can then extract the selected column
1.1.3. Compare the IDs of the 10 first reads. What are the differences between R1 and R2 IDs ?
zcat DATA/child.R1.fastq.gz | paste - - - - | cut -f 1 | head
zcat DATA/child.R2.fastq.gz | paste - - - - | cut -f 1 | head
bash1.1.4. What are the lengths of the Short-Reads ?
zcat DATA/child.R1.fastq.gz | paste - - - - | cut -f 2 | head
zcat DATA/child.R1.fastq.gz | paste - - - - | cut -f 2 | head -1 | tr -d '\n'| wc -c
bash1.1.5. Extract all the bases from the R1 reads of the child. Count the occurrences of each nucleotides.
zcat DATA/child.R1.fastq.gz | paste - - - - | cut -f 2 | head -100000 | grep -o . | sort | uniq -c
bashhere grep -o . means "show each character on a line"
head -100000 limits to 100,000 reads for faster processing
1.2. Quality Score
1.2.1. What are the character present in the score lines ?
zcat DATA/child.R1.fastq.gz | paste - - - - | cut -f 4 | head -100000 | grep -o . | sort | uniq | paste - - - - - -
bash- ASCII code from 33
!to 126~: 93 scores available

- Score = Phred
- Probability of incorrect identification:
Example: J ➡ code ASCII 74
1.3. Quality Control
Quality Control of the fastq files produced by the sequencer
- Quality preservation along the Reads
- Overall Quality of each Read
- GC% offset and base distribution might indicate contamination
fastqc
bashFile, Open : DATA / child.R1.gz
1.3.1. Good Per Base Quality

1.3.2. Bad Per Base Quality
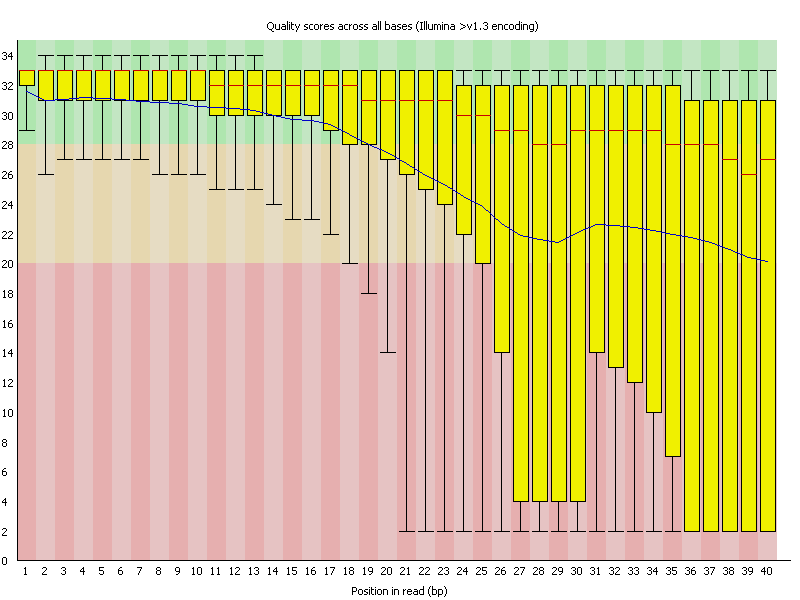
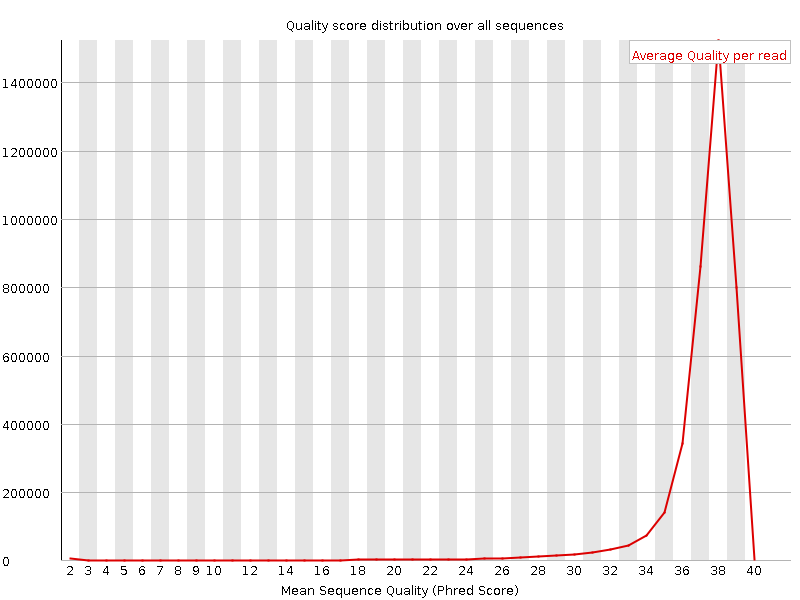
1.3.3. Good Per Sequence Quality
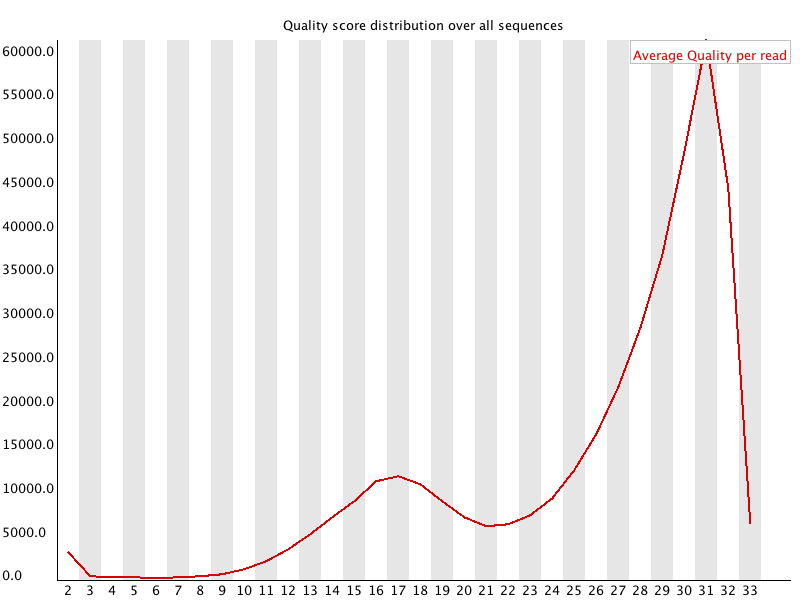
1.3.4. Bad Per Sequence Quality
1.3.5 mutiqc
Usually, we don't want to test each fastq file.
mkdir report
fastqc DATA/*fastq.gz --out report
multiqc report/*.zip
bashDownload the multiqc_report.html :
On Windows
- Refresh the file browse in mobaxterm
- Copy the
multiqc_report.htmlfile to windows - Open it
On Mac
open another terminal
type scp -P 4444 XXXXX@methionine:multiqc_report.html . (where XXXXX is your login)
Open the file
Next Step: 2 Alignments